

一、mcmctree树文件准备
1 树文件预处理
1.1 利用对前期获得的FigTree.tre文件进行根节点的调整
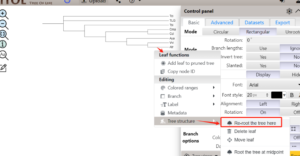
调整根节点,获得调整后的树文件
![]()
1.2 新建tree_deal.r,去掉除物种名以外的信息,获得如下树文件
#安装R包,如果安装失败,则直接输入R回车进入R程序,使用install.packages("ape") if(!requireNamespace("ape", quietly = TRUE)) install.packages("ape") library(ape) #修改输入的树文件名 MLtree <- read.tree("Tree_reroot.txt") #读取单拷贝进化 树 MLtree$edge.length <- NULL #删除分支长度信息 MLtree$node.label <- NULL write.tree(MLtree,"Tree_reroot.tree") #输出只有物种名>的进化树
1.3 执行tree_deal.r:新建do_tree_deal.sh并执行
#进入工作路径 cd /home/hztext/dai/Comp_Genome/Eight_ortho_result/Results_Nov20/Single_Copy_Orthologue_Sequences #运行处理树文件 Rscript /home/hztext/dai/Comp_Genome/Demo_deal/tree_deal.r
1.4 获得新的树文件Tree_reroot.tree
![]()
2 在 中查询已发表物种的分化时间
至少需要两个节点的时间,最好是在同一分枝的两个物种。
也可以在发表的文献中查找其分化时间。
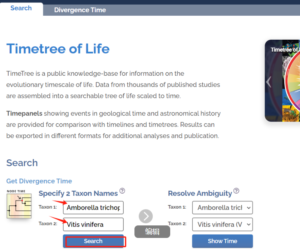
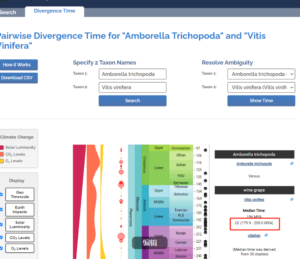
新建文件夹按照如下格式修改Tree_reroot.tree,8是物种数量,1不变
8 1
(Atr,(Vvi,(Aya,((Cma,Ccl)’>1.5<5.7′,(Tsi,(Tci,TLG)))))’>179.9<205.0′);
3 修改mcmctree的参数文件mcmctree.ctl,第一次运行选择usedata为3
seed = -1
seqfile = ./all.phy#单拷贝基因的phy格式
treefile = ./Tree_reroot.tree#包含了至少两个节点分化时间的进化树文件
outfile = out#输出文件不修改
ndata = 1 ## 必须注意为 1
seqtype = 2 * 0: nucleotides; 1:codons; 2:AAs ## 序列格式,蛋白就选择 AA
usedata = 3 * 0: no data; 1:seq like; 2:use in.BV; 3: out.BV ## 此步骤先设为 3
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates
RootAge = <450.0 * safe constraint on root age, used if no fossil for root.
model = 0 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
alpha = 0 * alpha for gamma rates at sites
ncatG = 5 * No. categories in discrete gamma
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
BDparas = 1 1 0 * birth, death, sampling
kappa_gamma = 6 2 * gamma prior for kappa
alpha_gamma = 1 1 * gamma prior for alpha
rgene_gamma = 2 2 * gamma prior for overall rates for genes
sigma2_gamma = 1 10 * gamma prior for sigma^2 (for clock=2 or 3)
finetune = 1: 0.1 0.1 0.1 0.01 .5 * auto (0 or 1) : times, musigma2, rates, mixing, paras, FossilErr
print = 1
burnin = 20000
sampfreq = 2
nsample = 100000
运行mcmctree.ctl
mcmctree mcmctree.ctl
4 复制out.BV、all.phy和Tree_reroot.tree到第二次运行mcmctree的文件夹
4.1 修改第二次mcmctree的运行参数,只需将usedata改为2即可
seed = -1
seqfile = ./all.phy
treefile = ./Tree_reroot.tree
outfile = out
ndata = 1 ## 必须注意为 1
seqtype = 2 * 0: nucleotides; 1:codons; 2:AAs ## 序列格式,蛋白就选择 AA
usedata = 2 * 0: no data; 1:seq like; 2:use in.BV; 3: out.BV
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates
RootAge = <450.0 * safe constraint on root age, used if no fossil for root.
model = 0 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
alpha = 0 * alpha for gamma rates at sites
ncatG = 5 * No. categories in discrete gamma
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
BDparas = 1 1 0 * birth, death, sampling
kappa_gamma = 6 2 * gamma prior for kappa
alpha_gamma = 1 1 * gamma prior for alpha
rgene_gamma = 2 2 * gamma prior for overall rates for genes
sigma2_gamma = 1 10 * gamma prior for sigma^2 (for clock=2 or 3)
finetune = 1: 0.1 0.1 0.1 0.01 .5 * auto (0 or 1) : times, musigma2, rates, mixing, paras, FossilErr
print = 1
burnin = 20000
sampfreq = 2
nsample = 100000
4.2 运行修改后的mcmctree.ctl
mcmctree mcmctree.ctl
4.3 最终获得的文件是FigTree.tre,比较两次的结果若无大差别,则正确
具体可以参考
![]()